



Przeprowadzono webinar na temat analizy ryzyka wyrobów medycznych zgodnie z wymaganiami ISO 13485 i ISO 14971.
Firma Med Quality przeprowadziła szkolenie na temat „Analiza ryzyka wyrobów medycznych zgodnie z wymaganiami ISO 13485…

Dyrektor Med Quality 2 kwietnia 2025 roku przeprowadził szkolenie dotyczące wymagań standardu ISO 13485:2016.
2 kwietnia 2025 roku dyrektor firmy Med Quality przeprowadził szkolenie dotyczące wymagań standardu ISO 13485:2016 dla…
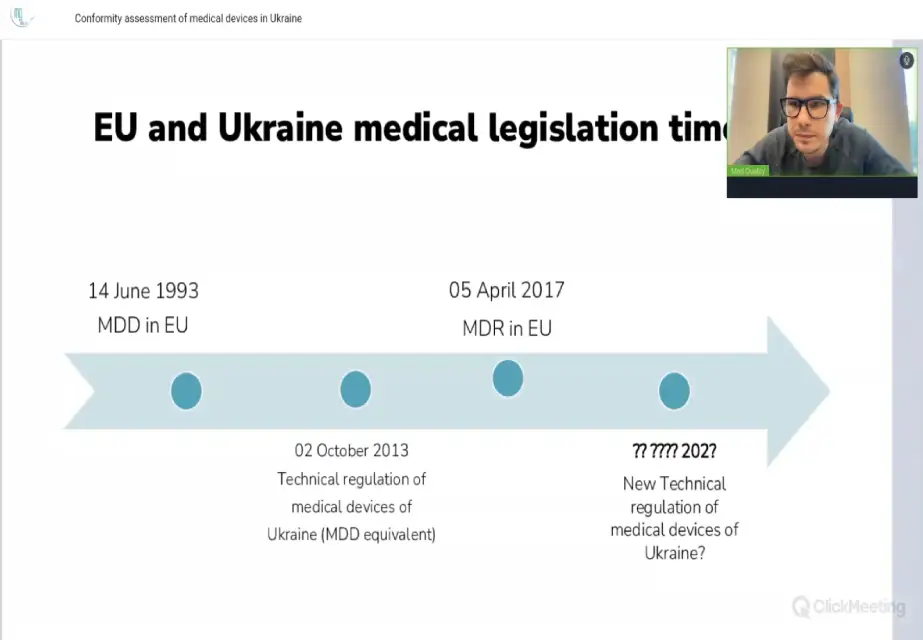
Dyrektor Med Quality Władysław Kasjanenko przeprowadził webinar na temat „Ocena zgodności wyrobów medycznych w Ukrainie”.
Prelegent omówił następujące tematy: Przegląd Technicznego Regulaminu Ukrainy nr 753 dotyczącego wyrobów medycznych: wprowadzenie, zakres stosowania…