



Директор Med Quality 2 квітня 2025 року провів навчання щодо вимог стандарту ISO 13485:2016
2 квітня 2025 року директор компанії Мед Кволіті провів навчання щодо вимог стандарту ISO 13485:2016 для…
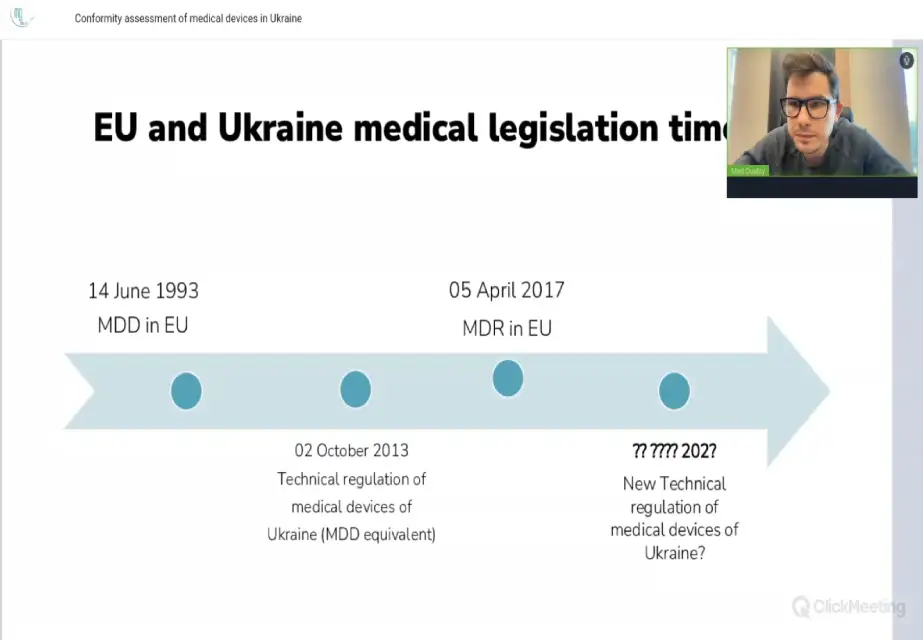
Директор Med Quality Владислав Касьяненко провів вебінар на тему «Оцінка відповідності медичних виробів в Україні»
Доповідач охопив такі теми: • Огляд Технічного регламенту України № 753 про медичні вироби: вступ, сфера…

Нещодавно компанія Med Quality провела вебінар на тему: "Як вийти на європейський ринок українському виробнику медичних виробів? Практичні поради"
На вебінарі спікером було розглянуто наступні питання: -Регулювання 2017/745 щодо медичних виробів, яке діє на території…